Recurrent
partial seizures with ictal yawning as atypical
presentation of Hashimoto's
encephalopathy
(steroid-responsive
encephalopathy associated with autoimmune
thyroiditis).
Casciato S, Di Bonaventura C, Lapenta L,
Fattouch J, Ferrazzano G, Fanella M, Di Fabio F,
Pasquini M, Amendolea MA, Manfredi M, Prencipe
M, Giallonardo AT.
Neurology Unit, Department
of Neuroscience, Sapienza University of Rome
Italy.
Hashimoto's encephalopathy (HE), also known
as steroid-responsive encephalopathy associated
with autoimmune thyroiditis (SREAT), is a rare
condition whose pathogenesis is unknown, though
autoimmune-mediated mechanisms are thought to be
involved. The prevalent neurological
manifestations of this disorder are epileptic
seizures and psychocognitive disorders
associated with EEG alterations. High
anti-thyroid antibody titers (particularly in
cerebrospinal fluid) and the effectiveness of
steroid therapy are usually considered to be
crucial elements in the diagnostic process. We
describe a 19-year-old female patient who had
been referred to the psychiatric unit because of
behavioral disorders characterized predominantly
by delirium with sexual content. She developed
recurrent focal seizures characterized by
atypical ictal semiology (repetitive forceful
yawning) and a rare EEG pattern (recurrent
seizures arising from the left temporal region
without evident "encephalopathic" activity). The
presence of anti-thyroperoxidase antibodies in
her cerebrospinal fluid and a good response to
steroids confirmed the diagnosis of HE. The
atypical presentation in the case we describe
appears to widen the electroclinical spectrum of
HE and highlights its importance for
differential diagnosis purposes in the
neuropsychiatric setting.
1. Introduction
Hashimoto's encephalopathy (HE), also called
steroid-responsive encephalopathy associated
with autoimmune thyroiditis (SREAT), is a rare
condition whose etiology is unknown, though
autoimmunemediated mechanisms are believed to be
involved in its pathogenesis [1, 2]. The
clinical onset of this condition is insidious or
acute, whereas its course is
relapsing&endash;remitting or monophasic in
nature, with varying neurological and, more
rarely, psychiatric symptoms (confusion,
seizures, myoclonus, personality changes,
psychosis, dementia, or strokelike episodes).
High anti-thyroid antibody titers (mainly
antithyroperoxidase [TPO-Ab], though
also anti-thyroglobulin [TG-Ab]) in
serum and cerebrospinal fluid (CSF) are the most
important diagnostic clues to the disease
[3, 4]. Neurological investigations do
not yield specific results, revealing, in the
majority of the cases, diffuse EEG
abnormalities, a moderately high CSF protein
content, and normal imaging. Current diagnostic
criteria include an excellent response to
corticosteroid therapy (observed in
approximately 50% of patients), which is
consequently the treatment of choice and results
in complete recovery [2].
We describe a 19-year-old woman with SREAT
whose first symptom at onset appears to have
been a primary behavior disorder. She developed
recurrent focal seizures characterized by an
atypical ictal semiology, that is, repetitive
yawning, associated with a rarely described EEG
pattern.
2. Case report
We describe a 19-year-old, right-handed
woman who has, since childhood, had
Graves&endash;Basedow disease, which has
responded well to methimazole therapy. One month
prior to admission to our department, the
patient developed highly distressing insomnia,
anxiety, depressed mood, poor motivation, and
social withdrawal, which were apparently due to
a recent stressful event. In the days following
hospitalization, she manifested an acute state
of psychomotor agitation and a delirious
syndrome, disinhibition, persecutory ideation,
incoherent speech, and psychotic disorder with
sexual content. She was admitted to the
psychiatric care unit so that the causes of the
primary psychotic disorder could be
investigated. The brain CT scan was normal and
routine blood tests did not reveal any
significant alterations; the patient was treated
with antipsychotic medication (olanzapine up to
10 mg/day), though with no significant
improvement. Some days later, she experienced
sudden brief episodes characterized by
repetitive forceful yawning, followed by
oroalimentary automatisms. Following speculation
regarding the possible presence of a
psycho-organic syndrome (after other toxic,
metabolic, and infectious causes of
encephalopathy had been excluded), the patient
was admitted to our neurological unit.
Her physical and neurological examinations
were unremarkable, if we exclude a lack of
initiative, bradykinesia, and mild postural
tremor in the left arm. The behavioral disorder
was characterized predominantly by akathisia,
disinhibition, and an excessive freehand drawing
tendency . The neuroimaging study (brain MRI
scan) did not disclose any significant
structural alterations (see below). No blood
count, electrolyte, clotting, or renal or
hepatic enzyme abnormalities were detected;
VDRL, TPHA, HIV, and serum vitamin B12 levels
were normal; autoimmune disease markers
(antinuclear antibody, rheumatoid factor level,
anti-double-stranded DNA antibody, anti-SS-A
antibody, anti-SS-B antibody, anticardiolipin
antibody, and myeloperoxidase anti-neutrophil
cytoplasmic antibody) were negative; thyroid
function tests disclosed a euthyreosis status
with high TG-Ab and TPO-Ab titers in serum
(1393.5 and 41.0 IU/mL, normal values: b100
IU/mL).
The CSF examination revealed a normal total
protein content (41 mg/dL), negative oligoclonal
bands, but high anti-thyroid antibody titers,
particularly those of TPO-Ab (1040.85 IU/mL,
normal level: 0) and TG-Ab (17.46 IU/mL, normal
level: 0); CSF findings also included a normal
Qalb (albumin CSF/serum quotient) that reflected
the integrity of the blood&endash;brain barrier
and the intrathecal synthesis of thyroid
autoantibodies. Serological and CSF screening
was performed to exclude toxic, metabolic,
infectious, and paraneoplastic etiologies.
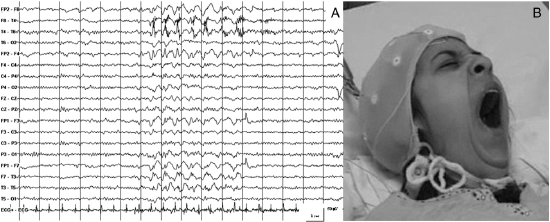
Video/EEG monitoring allowed us to record
several focal epileptic seizures. The EEG
patterns were characterized by rhythmic
monomorphic, mean-amplitude, 1.5- to 2-Hz,
slow-wave delta activity and, less frequently,
slow spike-and-wave complexes involving the left
temporal region and spreading over homologous
contralateral areas. The ictal clinical
semiology was characterized exclusively by
repetitive, stereotyped forceful yawning,
followed by oroalimentary automatisms (chewing)
and an inconstant unresponsive state.
Video/EEG examinations performed over time did
not yield any other clues (such as the presence
of a photoparoxysmal response) to help make a
diagnosis in this patient. The
neuropsychological evaluation was unfortunately
limited and incomplete because of the sedative
effect of drugs (administered for the behavioral
disorders) and the poor level of collaboration
offered by the patient herself. However, an
interesting and curious finding emerged from the
Rey&endash;Osterrieth Complex Figure Test,
which, when occasionally performed during the
peri-ictal phase, revealed a transient
impairment that was closely related to the
seizure activity of the specific performances. A
further MRI scan revealed mild unspecific
periventricular white matter hyperintensities on
fluid-attenuated inversion&endash;recovery
(FLAIR) and T2-weighted images.
In the acute phase, high-dose intravenous
steroid therapy was started (methylprednisolone
at 1000 mg daily for 5 days), followed by an
oral formulation (dexamethasone at an initial
daily dose of 1 mg/kg). Steroid therapy led to
progressive improvement in the patient. The
epileptic seizures promptly disappeared and the
psychiatric manifestations resolved completely
within a few days. Periodic follow-up EEGs
revealed normal background activity without
focal or diffuse epileptiform discharges.
Steroids were maintained for almost 6 months
(0.5 mg/day), after which they were gradually
suspended. Antipsychotic medications were
tapered and withdrawn after 2 weeks. During an
8-month follow-up, the patient did not
experience any additional seizures and her
psychocognitive disorders fully resolved. At the
last visit, the patient did not need any therapy
and had regained her ability to perform all her
daily activities (including her studies).
3. Discussion
Hashimoto's encephalopathy, also referred to
as SREAT, is a rare condition whose etiology is
unknown, though autoimmune-mediated mechanisms
are believed to be involved in its pathogenesis
[1, 2]. The pathogenetic hypotheses
include autoimmune vasculitis, autoimmune
reaction to antigens shared by the thyroid gland
and the central nervous system, cerebral
hypoperfusion, and the toxic effect of
thyrotropin- releasing hormone [4, 5].
To date, HE is described as a disease affecting
both the neuroendocrine and nervous systems.
Existing pathological reports have demonstrated
the presence of gliosis and mild lymphocyte
infiltration in brain parenchyma around small
vessels [6&endash;8]. The
pathophysiology includes brain microvascular
inflammation resulting from immune complex
deposition and autoimmune mediated thyroiditis.
Thyroid antibodies or unknown anti-neuron
antibodies may lead to damage in both thyroid
and neural tissue [8, 9].
Misdiagnosis of HE at presentation is
common, while diagnosis is based above all on
the exclusion of more common diseases owing to
the variability of the symptoms [1, 5].
Peschen-Rosin and co-workers [10] first
suggested that HE could be diagnosed on the
basis of the unexplained occurrence of relapsing
myoclonus, generalized or focal seizures,
psychiatric disorders, or focal neurological
deficits associated with three of the following
conditions: abnormal EEG, high thyroid
autoantibodies, high CSF protein, excellent
response to steroids, or unrevealing cerebral
MRI [2, 11]. The range of clinical
symptoms and signs has, since this early
proposal, been extended considerably, whereas
the response to steroids has been shown to vary
substantially. Some authors have also suggested
that, besides high serum levels, high CSF
anti-thyroid antibody titers may be more
pathognomonic [4] than responsiveness to
corticosteroid therapy, which occurs in only 50%
of patients [10]. Other authors have
instead recently supported responsiveness to
corticosteroid therapy as a means of diagnosing
HE [11]. In 2006, Ferracci and Carnevale
[3] summarized the diagnostic principles
followed by the majority of authors: after
ruling out other diseases, high serum
anti-thyroid antibody levels may be considered
to support the diagnosis of HE, but should not
be considered as definitive proof of this
disease.
With respect to immunological aspects, this
case seems to relaunch the crucial role of
dysimmune mechanisms in some rare epilepsy
conditions that are clinically characterized by
an association between seizures and
psychocognitive disturbances. These conditions
usually include systemic autoimmune diseases
with secondary brain involvement (such as
systemic lupus erythematosus, Sjogren's
syndrome, anti-phospholipid syndrome, systemic
vasculitis, and coeliac disease) [12]
and autoimmune encephalitis electively affecting
the central nervous system (paraneoplastic and
non-paraneoplastic limbic encephalitis,
encephalitis associated with VGKC-complex-Ab ,
anti-NMDARs, AMPARs-Ab, GABA(B)Rs-Ab, GlyRs-Ab,
and GAD-Ab) [13, 14]. Although we had
contemplated other autoimmune steroidresponsive
encephalopathies in our case, the serum and CSF
findings (high serum and CSF levels of TPO-Ab,
normal Qalb pointing to the integrity of the
blood&endash;brain barrier and confirming the
presence of intrathecal synthesis of these
autoantibodies), the responsiveness to steroids,
and the patient's medical history supported a
diagnosis of HE. The normal titers of the other
autoantibodies allowed us to rule out the
aforementioned systemic conditions. As regards
pathologies that more specifically affect the
central nervous system, we decided not to
perform the complete autoimmune screening (i.e.,
VGKCcomplex- Ab, antiNMDARs, AMPARs-Ab,
GABA(B)Rs-Ab, GlyRs-Ab, and GAD-Ab), though we
recognize that it should have been conducted
according to a modern approach to seizure
disorders of unknown origin.
The case we describe may be considered of
considerable interest for several reasons. From
an electroclinical point of view, the
repetitive, stereotyped yawning observed in
our patient as the main, unusual ictal clinical
manifestation during recurrent partial seizures
arising from the left temporal regions and
spreading to the contralateral areas is
particularly worthy of note. The occurrence
of yawning in relation to epileptic seizures
has rarely been described in the clinical
setting [15&endash;21]. Neural
structures underlying this complex
spatiotemporal physiological reflex are
presumably located in the brainstem. Different
mechanisms might explain peri-ictal
yawning, including direct activation of the
brainstem structures by the epileptic discharge,
seizure-mediated secretion of endogenous
neurohumoral substances (such as prolactin and
oxytocin), and functional changes of the
brainstem related to the level of alertness. As
it has usually been reported in seizures
involving primarily the nondominant hemisphere,
some authors have attributed a lateralizing
value to this phenomenon [15, 16]. In
contrast to this observation, though in
agreement with other articles reporting left
temporal or bitemporal ictal discharges [18,
21&endash;23], the seizures in our case
appeared to involve primarily the left
hemisphere.
An EEG corresponding to forceful yawning
and consisting of a focal ictal pattern has also
rarely been observed in this syndrome. Yet
another interesting finding is that the EEG
tracing did not, if we exclude the recurrent
seizures, reveal any "encephalopathic" pattern,
but instead showed normal background activity.
The most frequently reported abnormal patterns
in the few studies that have focused on EEG
findings in HE [3, 17, 22, 24] are: (1)
slowing, continuous or intermittent background
activity, often associated with diffuse,
highamplitude, theta&endash;delta rhythms
[3, 22, 24] and (2) epileptic ictal
activity, sometimes structured as focal or
generalized status epilepticus [8, 17,
23].
From a psychocognitive point of view, the
psychiatric manifestations of HE in our case
were characterized by delirium with sexual
content and compulsive drawing and writing,
which were similar to and could easily be
misdiagnosed as the typical onset of primary
psychotic disorder. However, the combination of
a poor response to antipsychotic agents,
epileptic seizures, EEG alterations, and a
surprisingly prompt response to steroids
(worsening of the psychiatric manifestations
might have been expected instead) supported, in
our case, the hypothesis of a psycho-organic
condition and oriented the diagnostic process.
With respect to the cognitive and behavioral
aspects of HE, some alterations are secondary to
the encephalopathy itself and to recurrent
seizures. Indeed, seizures, which occasionally
occurred during the neuropsychological
evaluation in our patient, produced a further
transient worsening in the cognitive
deficits.
In keeping with published reports, according
to which normal neuroimaging or nonspecific
white matter abnormalities are the most common
imaging findings in HE [1, 2, 25], the
MRI scan obtained did not disclose any
significant structural abnormality. Zhao and
colleagues [26] recently described
atypical neuroimaging changes in a patient with
HE, consisting of multifocal abnormalities in
cortical and subcortical areas not enhanced by
gadolinium that rapidly resolved, together with
the clinical symptoms, after high doses of
corticosteroid therapy. In our patient, the
SPECT scan revealed a focal area of
hypoperfusion involving mainly the left temporal
regions and other areas of abnormal perfusion in
the homolateral frontal and parietal lobes; this
neuroimaging pattern was highly concordant with
the electroclinical findings.
In conclusion, the case we describe not only
appears to widen the electroclinical patterns
associated with HE, thereby confirming the fact
that our knowledge of this disease remains
incomplete, but also lends further support to
both the central role of anti-thyroid antibodies
in the CSF for diagnostic purposes and the
effectiveness of steroid therapy. Within the
context of a differential diagnosis, psychiatric
disorders associated with epileptic seizures
with atypical features, particularly in patients
with a history of autoimmune thyroiditis, should
always raise suspicions for this condition so
that prompt and effective treatment may be
provided.